Un enorme árbol genealógico para comprender la familia humana
Combinar miles de genomas modernos y antiguos para construir la mayor genealogía humana hasta la fecha es lo que ha logrado un equipo internacional de científicos. El estudio, publicado en la revista Science, muestra los principales acontecimientos de la historia de la humanidad, así como su cronología y localización geográfica.
La secuenciación del genoma humano en las dos últimas décadas ha dado lugar a un conocimiento más profundo de nuestro pasado evolutivo. De esta forma, se han generado datos genómicos de cientos de miles de individuos, incluidos los de miles de personas prehistóricas.
Un equipo de científicos ha aplicado un nuevo método no paramétrico que combina datos de registro en árbol de genomas humanos antiguos y modernos, lo que les ha permitido deducir una genealogía completa humana.
“Un método no paramétrico significa que tuvimos que hacer muy pocas suposiciones sobre la naturaleza de las migraciones humanas. Por ejemplo, no necesitamos conjeturar si hubo una, o solo unas pocas migraciones fuera de África, o que ocurrieron de una manera determinada, en un momento concreto. Nuestro objetivo es dejar que los datos hablen por sí mismos”, dice a SINC Yan Wong, del Centro Li Ka Shing para la Información y el Descubrimiento de la Salud en la Universidad de Oxford (Reino Unido) y coautor del estudio que publica la revista Science.
Con esta técnica, el ancestro de dos individuos se asigna geográficamente al punto medio entre la localización geográfica de sus dos descendientes y se van determinando sus ancestros en el pasado. “Aunque sabemos que este método es imperfecto, parece recapitular bien muchos de los movimientos humanos conocidos. Quizás la sorpresa es, de hecho, que funciona razonablemente bien”, declara a SINC Aida Andres, del Instituto de Genética del University College de Londres y autora de un artículo en la misma revista, como comentario a este trabajo.
Hasta la fecha, se habían recogido miles de genomas humanos, que contienen segmentos de diferentes y múltiples ancestros de distintas edades. En consecuencia, la elaboración de una imagen completa de la genealogía y la variación genómica a lo largo de la historia humana representa un reto técnico.
AHora, Wong y su equipo han logrado construir un enorme árbol genealógico para toda la humanidad. “Al tratar toda nuestra ascendencia como una sola red, podemos estimar características generales de los antepasados comunes en el árbol genealógico humano, como su edad e incluso potencialmente su ubicación. Nuestro método se podrá escalar potencialmente a millones de genomas”, asegura Wong.
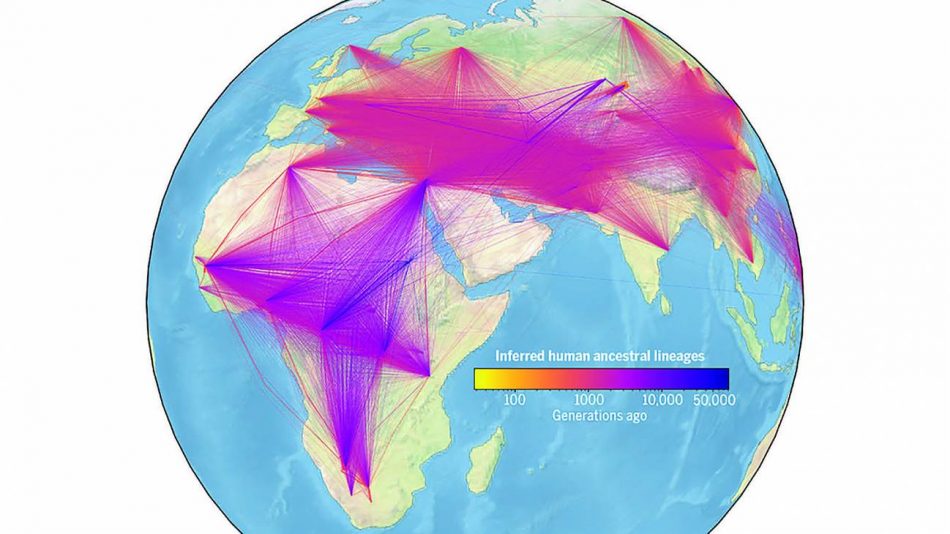
Una genealogía unificada de los genomas modernos y antiguos. / Wilder Wohns
La historia que generó toda nuestra variación genética
Dado que las regiones genómicas individuales solo se heredan de un progenitor, la ascendencia de cada punto del genoma puede considerarse como un árbol. El conjunto de árboles, conocido como ‘secuencia arbórea’ o ‘gráfico de recombinación ancestral’, enlaza las regiones genéticas a través del tiempo con los ancestros en los que apareció por primera vez la variación genética.
En total, se usaron ocho bases de datos diferentes que incluyeron un total de 3.609 secuencias genómicas individuales de 215 poblaciones. Los genomas antiguos incluían muestras halladas en todo el mundo, con edades comprendidas entre 1.000 y más de 100.000 años. Los algoritmos predijeron dónde debían estar presentes los ancestros comunes en los árboles evolutivos para explicar los patrones de variación genética. La red resultante contenía casi 27 millones de ancestros.
Anthony Wilder Wohns, que llevó a cabo la investigación en la Universidad de Oxford y que ahora es investigador posdoctoral en el Instituto Broad del MIT y Harvard (EE UU), apunta a SINC: “Aunque en esta genealogía se incluye una enorme cantidad de detalles, observar la edad media y la ubicación de los antepasados proporciona una gran visión de las características generales de la historia humana. A veces, podemos incluso mostrar todos estos datos para revelar patrones importantes”.
Tras añadir los datos de localización de estos genomas de muestra, los autores utilizaron la red para estimar dónde habían vivido los ancestros comunes. Los resultados recapitularon con éxito acontecimientos clave de la historia evolutiva humana, incluida la migración fuera de África.
“Hace tiempo que se sabe que hubo una dispersión fuera de este continente, quizá hace unos 100.000 años. Las señales de este acontecimiento se encuentran en partes del genoma como el mitocondrial, el cromosoma Y y varios otros genes. Sin embargo, nuestra genealogía muestra, por primera vez, que la señal de este acontecimiento está claramente presente en todo el genoma”, señala Wong.
Los investigadores observaron señales de linajes ancestrales muy profundos en África, el evento fuera de África, y la introgresión o incorporación arcaica de genes en Oceanía.
Las mutaciones que nos dan las pistas
Este método también tiene en cuenta los datos que faltan y los erróneos, y utiliza genomas antiguos fragmentados para ayudar a identificar el momento de la aparición de los alelos.
“Estos son variantes genéticas. Aparecen por mutación en algún momento, y si se establecen a partir de ese instante, esa posición genómica será variable en la población. Unos cromosomas tendrán un alelo, otros el otro. Podemos inferir la edad de esos alelos usando genomas modernos, pero es un problema difícil y lo hacemos con poca resolución”, revela Andres.
Lo que han hecho en este estudio es utilizar genomas antiguos de baja calidad para ayudar a determinar su edad. Para cada alelo se han preguntado cuándo lo ven por primera vez. Si, por ejemplo, se observa en un genoma de hace 5.000 años, sabrán con certeza que esa mutación tiene más de esa edad.
“Esto ayuda a mejorar los modelos que nos permiten inferir la historia demográfica humana. Pero, además, si ese alelo tiene efectos importantes —por ejemplo, si nos permite digerir la lactosa, o si aumenta el riesgo de una enfermedad—, mejorar la inferencia de la edad del alelo nos ayuda a entender la historia de ese fenotipo o de esa enfermedad”, continúa la investigadora del University College.
Gil McVean, otro de los coautores de la Universidad de Oxford, recalca a SINC: “Los conjuntos de datos genómicos que utilizamos se han construido a partir de muchas fuentes diferentes y utilizando distintos métodos. Inevitablemente, se producen ciertos tipos de error. Nuestro enfoque ayuda a identificarlos. Estimamos que la tasa es pequeña, menos del 0,5 % de los lugares de variantes en el genoma, pero su eliminación crea una imagen más precisa y completa de la variación genómica humana”.
Aunque el estudio se centra en los seres humanos, el método es válido para la mayoría de los seres vivos.
Limitaciones del estudio
Wilder Wohns, por su parte, explica que una de las principales limitaciones de este trabajo es que utilizan un método muy simple para estimar la ubicación de nuestros antepasados.
“Se podría trabajar mucho más en este campo. Además, nuestras estimaciones de la ubicación de los ancestros están limitadas en última instancia por las de las secuencias genómicas. Por ejemplo, la precisión de nuestra reconstrucción de las migraciones de los pueblos indígenas a las Américas se ve dificultada por la relativa escasez de muestras del noreste de Siberia y el noroeste de Norteamérica”, añade.
Además, si se produjeran grandes migraciones históricas que no dejaran descendientes locales, la precisión de dichas estimaciones de la ubicación de los ancestros se vería disminuida.
Por último, el método tiene en cuenta los errores de los conjuntos de datos genéticos utilizados, pero no lo hace a la perfección. Esto también puede afectar a las estimaciones de la edad y la ubicación de nuestros antepasados.
“Desde mi punto de vista, una de las mayores limitaciones de todo estudio que utiliza las grandes bases de datos de genomas existentes —incluido este— es que no tenemos una representación adecuada de todas las poblaciones humanas. Las bases de datos están sesgadas hacia poblaciones muy estudiadas. Por ejemplo, las europeas. Pero esto ocurre, no solo en esta investigación, sino en la mayoría del trabajo genómico actual, y se solucionará solo secuenciando más genomas de más poblaciones mundiales”, concluye Andres.
Referencia:
Anthony Wilder Wohns et al. «A unified genealogy of modern and ancient genomes». Science
Relacionado
